Data-driven approaches have experienced an impressive growth over the last few years, thanks to the availability of large resources (i.e., databases) and the development of off-the-shelf machine learning (ML) methods. The combination of big data and ML is sparking the “fourth industrial revolution” and a new paradigm of science. In this context, it can add a new dimension to chemical discovery and allow to accelerate the chemicals value chain from discovery to deployment. Many flavours of ML are available, among which two main types stand out, namely unsupervised and supervised machine learning. Both types of approaches can provide great insights and be applied at different stages of the discovery of new chemicals.
Unsupervised learning methods use unlabelled data to discover structure and pattern information via different types of approaches such as:
- Dimensionality reduction methods, e.g. principal component analysis (PCA), which offer ways to reduce the dimensionality of the highly multidimensional chemical space to a 2D or 3D space that can be properly visualized.
- Clustering algorithms, e.g. K-Means or Ward clustering, which detect patterns within the input data and therefore permit to identify groups of molecules with similar features.
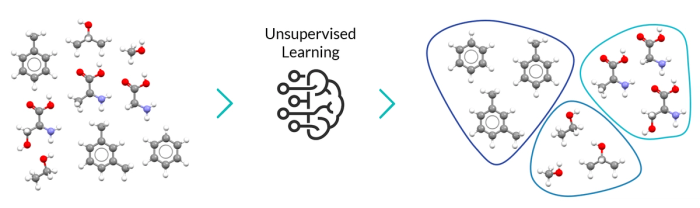
These methods allow to visualize and understand the relevant chemical space of a given problem to tackle. In particular, the identification of groups of molecules with similar features can be an important driver of the discovery process, enabling educated prioritization decisions. For example, one can first decide to broadly sample the chemical space, either via experimental measurements or in-silico molecular modelling, and then focus on the space around hits to further optimize the performance of the chemicals.
Once a number of laboratory or in-silico modelling experiments have been performed, supervised learning algorithms can be leveraged to build predictive models based on both input data and labelled results.
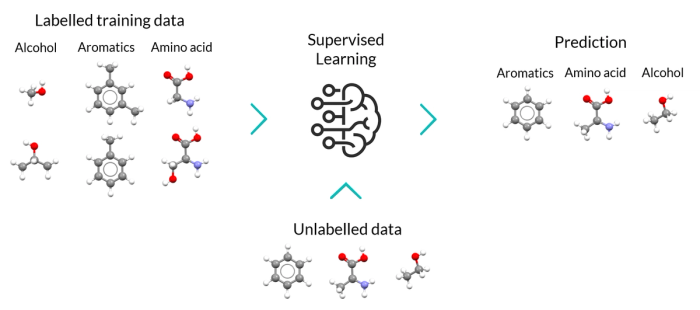
Supervised learning problems can be grouped into two types of problems:
- Classification: when the experimental result can be categorized such as “black” or “white”. Classifications are not restricted to two-class problems, but the balance of the dataset is an important aspect to consider.
- Regression: when the experimental result is a real value such “interatomic distance” or “kinetic energy”.
Often, a problem can be tackled from both angles (classification and regression) by defining threshold values. For example, one could try to predict the pH of a compound or its acidic/neutral/alkaline character.
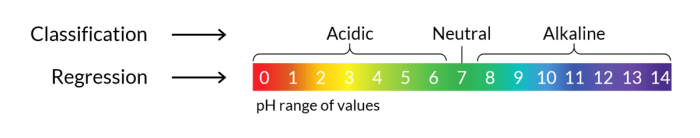
Conversely to unsupervised approaches, these models actually allow to predict properties and to identify the most promising candidate compounds. Therefore, the testing of such promising candidates, either computationally or experimentally, can be prioritized, providing a tremendous acceleration of the discovery process. These models are also particularly powerful to identify structure-activity relationships.
Contact us to explore how data-driven approaches can help in your chemical R&D.