A predominant challenge for the oil and gas industry is the deposition of asphaltene, a class of compounds defined as the fraction of crude oil that is soluble in toluene but not in n-heptane. Asphaltene, known as the “cholesterol” of crude oil, is among the heaviest and most polar components of petroleum fluids and can precipitate in the reservoir and plug production and transportation flowlines, risking economic loss due to flow interruption and environmental damages. An efficient and economical mitigation strategy consists of injecting chemical additives, referred to as asphaltene inhibitors, to stabilize asphaltene in crude oil. Hence, the development of these additives is of high industrial relevance but is often hindered because of the oil field dependence of asphaltene stability and aggregation behavior. Therefore, it seems critical to understand and rationalize the underlying mechanisms of the asphaltene aggregation process to better address its inhibition.
Generally described as a very polydisperse class of organic solids made of a variety of polyaromatic structures with aliphatic chains or heteroatoms, either organic or metallic the precise molecular structure of asphaltene has been a longstanding subject of debate and an important focus during decades of investigation. To account for asphaltene molecular polydispersity while making the large combinatorial space of possible asphaltene blends accessible to a systematic study via simulation, Nextmol has implemented an upfront unsupervised machine-learning approach (clustering) to identify a reduced set of 6 model molecules representative of the diversity of asphaltene among a catalogue of 89 model molecules.
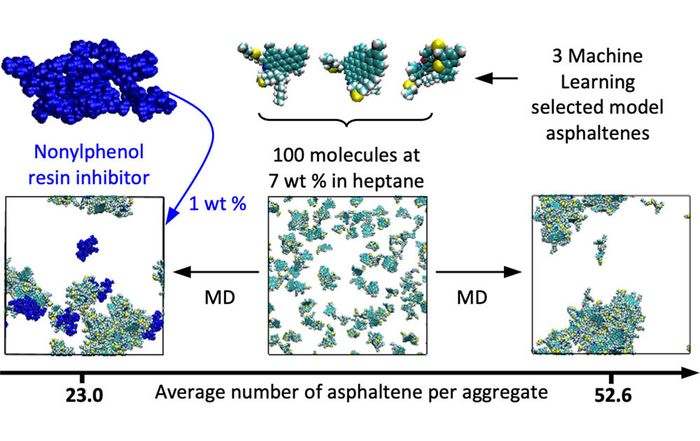
Molecular dynamics (MD) simulation protocols have been optimized and a series of analysis observables have been implemented to permit the investigation of asphaltene aggregation in toluene and heptane. The aggregation number, which corresponds to the average number of molecules per aggregate, permits monitoring the equilibrium state of the system while estimating the size of asphaltene aggregates. Further, the size of these aggregates can also be characterized via their radius of gyration, while an estimate of their density and their relative shape anisotropy provide information relative to their shape. Moreover, to enable a deeper understanding of the dynamics of the aggregation process and in particular the formation of aggregates in mixture simulations, we have investigated the detailed composition of aggregates at different points of the trajectories and have monitored the percentage of each type of asphaltene molecule participating in the formation of aggregates along the trajectory.
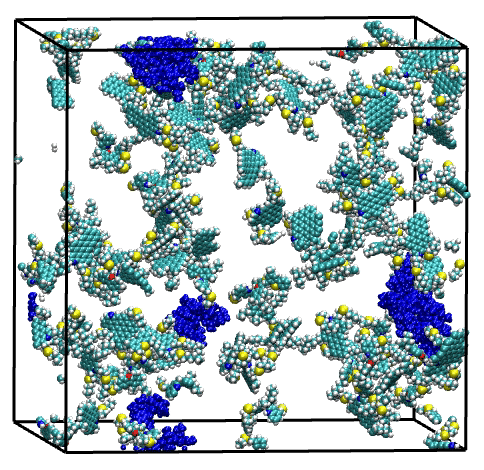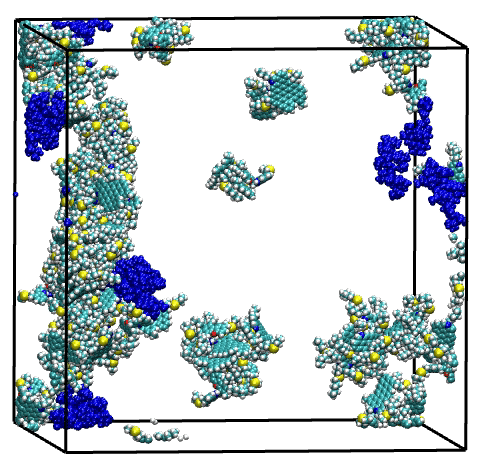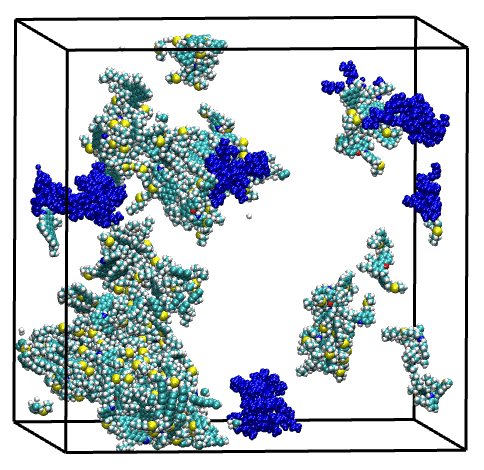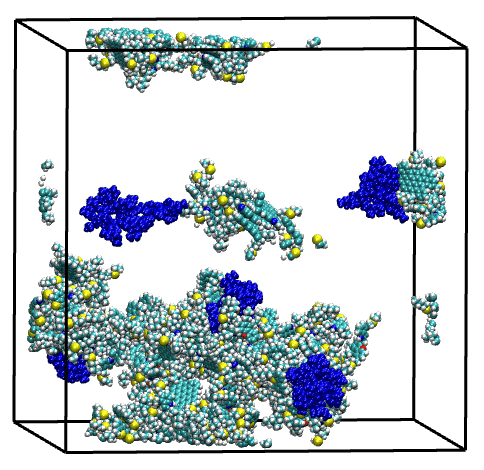
Overall, single asphaltene model simulations have shown a broad range of aggregation behaviors, driven by structural features such as size of the aromatic core, length of the aliphatic chains, and presence of heteroatoms. Then, the combination of these model molecules in a series of mixtures have highlighted the complex and diverse effects of molecular polydispersity on the aggregation process of asphaltene. Simulations revealed both antagonistic and synergistic effects mediated by the trigger or facilitator action of specific asphaltene model molecules. These findings illustrate the necessity of accounting for molecular polydispersity when studying the asphaltene aggregation process.
Last but not least, the suitability of the MD simulation and analysis workflow for the evaluation of asphaltene inhibitor performance has been demonstrated through a showcase for a nonylphenol resin inhibitor.
All the details can be found in our paper in collaboration with Clariant Oil Services and published open access in ACS Omega. Contact us for more information.